
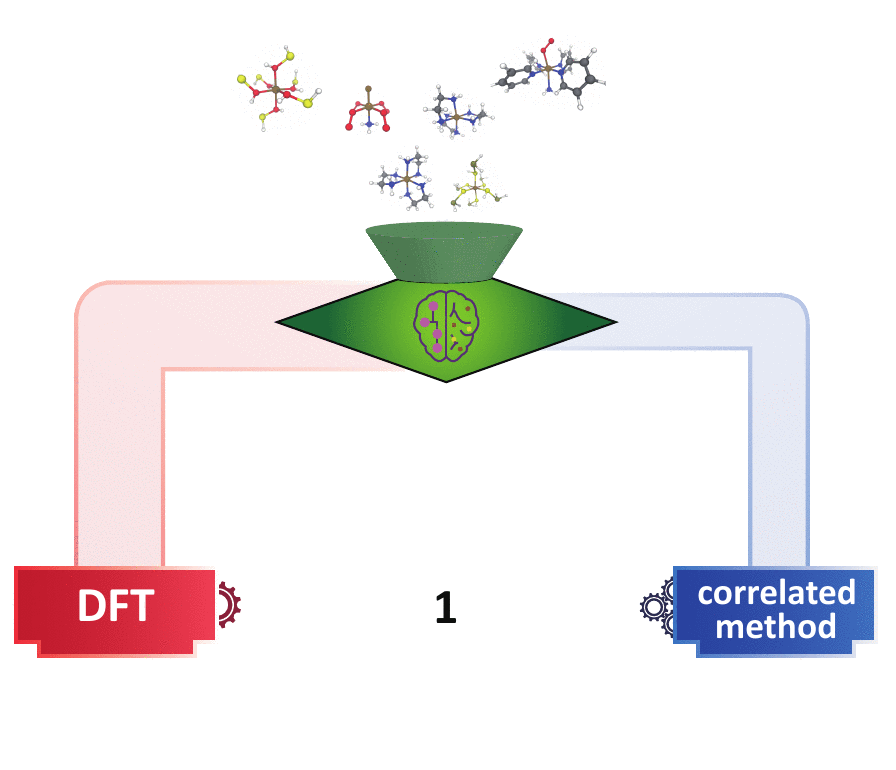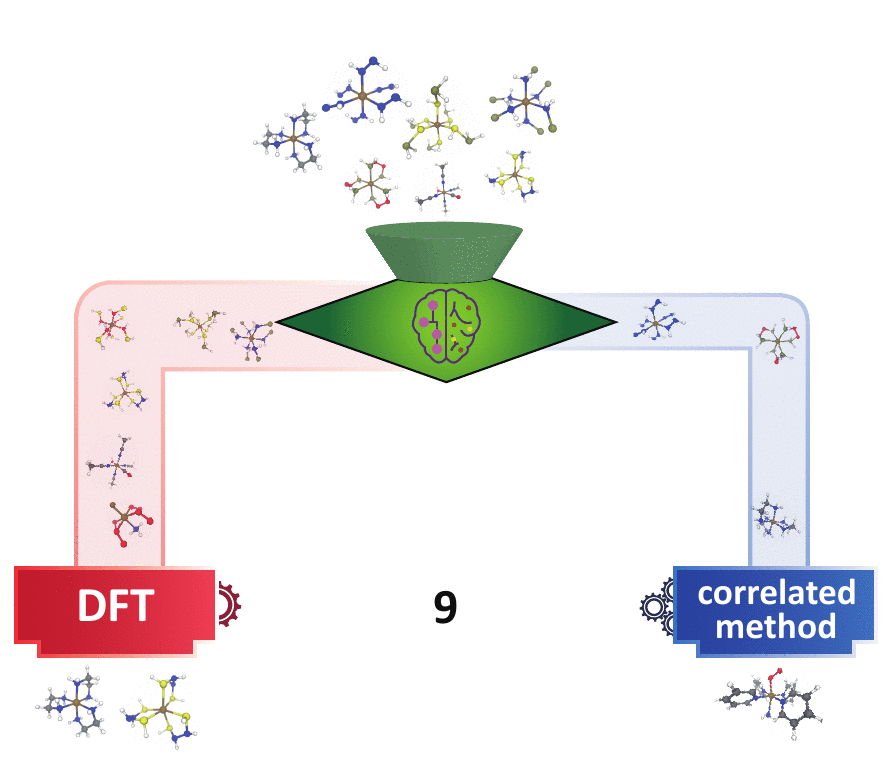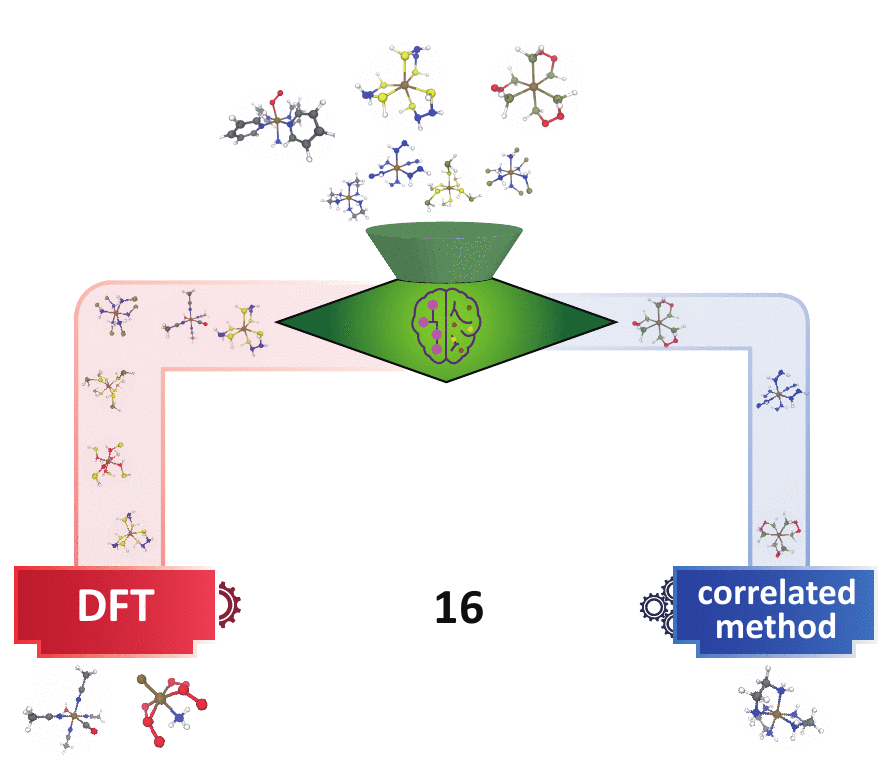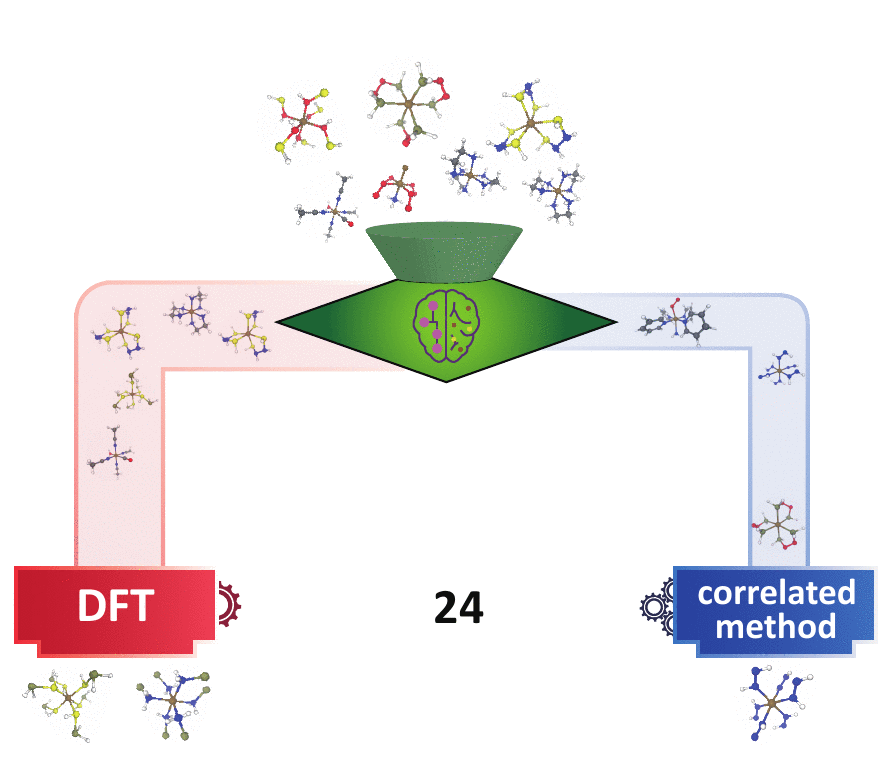

decision engine

Overcoming cost-accuracy trade off in computational chemistry with ML-assisted method selections
Chem. Sci. 13, 4962 (2022)
J. Phys. Chem. Lett. 11, 16, 6640–6648 (2020)
J. Chem. Theory Comput. 16, 7, 4373–4387 (2020)
Many experts have devised multi-reference (MR) diagnostics to detect when correlated wavefunction theory (WFT) is needed over DFT, but these tools are impractical in large-scale materials discovery, because they either request expensive calculations or the diagnostics themselves may behave unpredictably in new materials spaces. I first bridged the gap of the cost-accuracy tradeoff between DFT- and WFT-based MR diagnostics by building regression models to accurately predict those WFT-based diagnostics using DFT-based diagnostics and the 3D geometry of a system as inputs. In addition, utilizing the underlying consensus among multiple MR diagnostics,
I developed a semi-supervised learning model that classifies based on both the labeled points that contain extremely strong/weak MR diagnostics and the underlying distributions of the MR diagnostics of all points. This model far outperforms the existing unsupervised learning (i.e., clustering) methods and conventional cutoff-based approach widely used in the chemistry community in distinguishing systems that contain strong MR character. This model is readily transferable to larger systems with unseen chemical compositions. Combining with our previously built regression models, one can obtain faithful MR character classification at a low cost of DFT. This set of ML models pave the way for quickly identifying the “DFT-safe” island6 during materials discovery and subsequently performing higher-cost correlated WFT calculations only for systems that are promising but difficult for DFT.